As the fundamental cellular unit responsible for making the heart beat, cardiomyocytes are fundamental in cardiovascular research. Stress or damage to the cells – for example following ischemia – has profound effects on cardiac muscle cells, which can be assessed in vitro, providing valuable insights into the diseased state.
The following two articles, published in Science Journals and Nature Communications, respectively, highlight the current use of Aurora Scientific instrumentation to study calcium-activated force production in skinned cardiac myocytes.
Disruption of cardiac thin filament assembly arising from a mutation in LMOD2: A novel mechanism of neonatal dilated cardiomyopathy
As our knowledge and ability to perform genetic sequencing and analysis has improved, the role of genetics in heart disease has become increasingly apparent. Especially, as the authors note, in pediatric dilated cardiomyopathy (DCM), in which pathogenic mutations have been identified in 40% of all patients tested.
In this paper, Ahrens-Nicklas et. al. describe a unique opportunity to study a previously-unseen form of neonatal DCM, in which the patient has a biallelic homozygous nonsense variant in leiomodin 2 (LMOD2), an actin-binding protein essential for thin filament assembly in heart cells. Due to the severity of the phenotype, the infant was the recipient of a heart transplant at 10 months of age and allowed the researchers to study the explanted heart alongside Lmod2-null mice to assess the mutation’s effects on cardiac function.
The researchers found that the heart cells had exceptionally short thin filaments, which are critical for force production in muscle cells. Indeed, the researchers used the Aurora Scientific 803B permeabilized myocyte apparatus along with the 406A force transducer to assess force production in the cardiac myocytes. They found that, in both the patient’s heart cells and cells from Lmod2-null mice, maximum force production was severely reduced compared to healthy samples. In addition, calcium sensitivity was decreased.
The authors conclude the paper by noting that while theirs was the first recorded case of this biallelic homozygous genotype, it is very likely that previous cases were simply undiagnosed, given the historical lack of genetic sequencing.
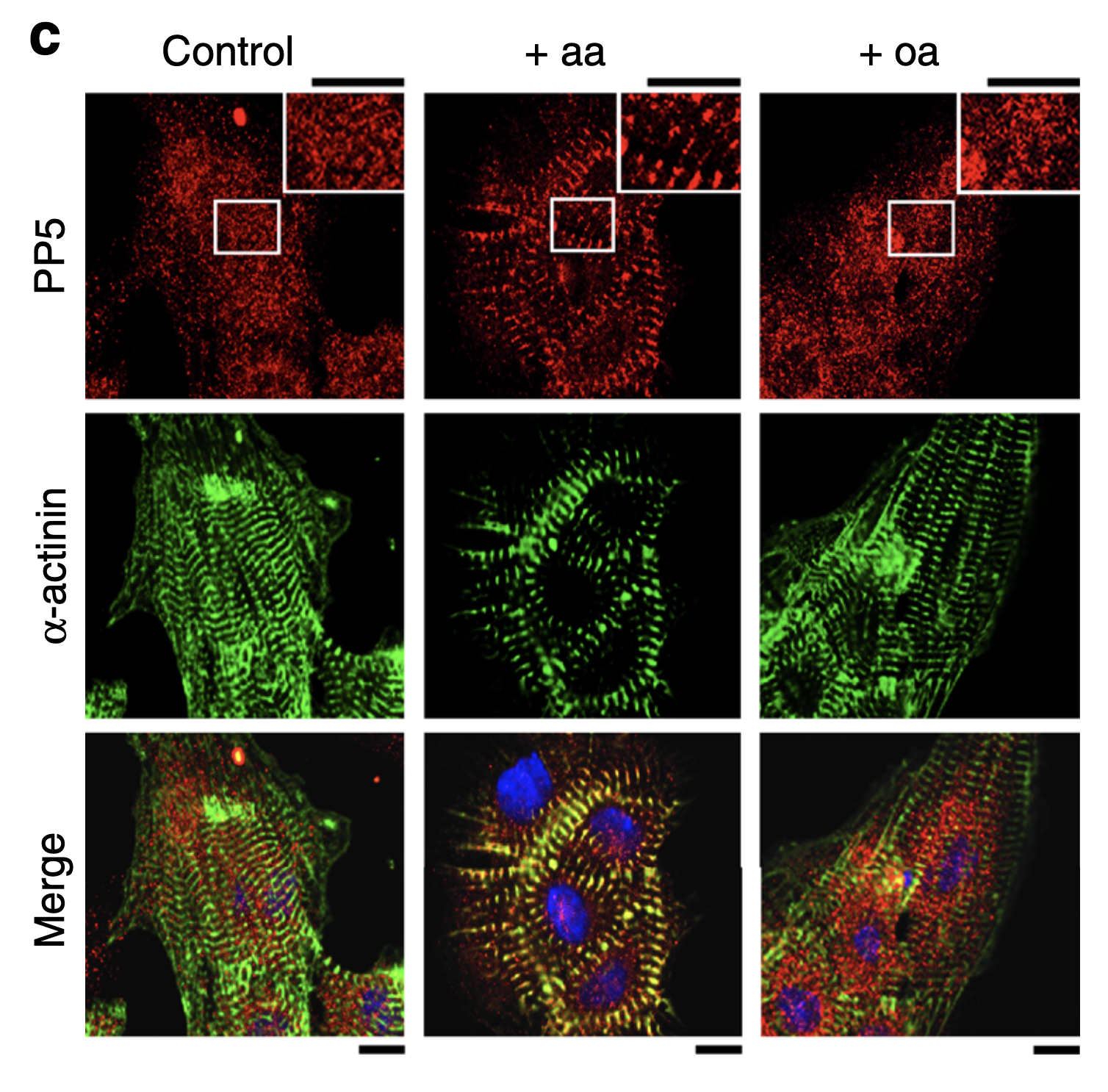
Protein phosphatase 5 regulates titin phosphorylation and function at a sarcomere-associated mechanosensor complex
Many proteins must come together to maintain functional flexibility and contraction in sarcomeres. Some of these proteins contain sequences unique to the heart which can interact with kinases and phosphatases to quickly respond to the body’s demands, for example during exercise.
In this study, Krysiak et. al. found that protein phosphatase 5 (PP5), which is ubiquitously expressed in eukaryotic cells, interacts with titin in cardiac myocytes. Previously, the function of PP5 in cardiomyocytes was unknown. Specifically, they determined that PP5 dephosphorylates the N2B unique sequence (N2Bus), the only sequence in Titin unique to cardiomyocyte-specific isoforms.
The N2Bus is an important site – it functions as a protein-protein hub, serves as a key phosphorylation site and acts as an elastic spring element, helping to maintain passive tension. Phosphorylation of the sequence lowers passive tension of the cardiomyocyte. In addition, in both human disease and disease models, N2B is often under-phosphorylated, contributing to overly stiff heart tissue.
To assess PP5’s interactions with titin and its role in heart disease, they used a suite of tests, including assays looking at phosphorylation and protein binding, cell culture manipulation and transgenic mouse models. Using the Aurora Scientific 1600A Permeabilized Myocyte Test System, they found that PP5 antagonize kinases that lower titin-based passive tension.
In addition, they found that PP5 dephosphorylates titin at the N2Bus, antagonizes kinases that phosphorylate it, and is over-expressed during heart disease. They note that this may explain why N2Bus is hypo-phosphorylated in heart disease. They conclude, that:
“PP5 is an antagonist to the various PKs that phosphorylate N2Bus, in terms of its effect on cardiomyocyte Fpassive” and “reversing the induction of PP5 in heart failure would promote titin phosphorylation and help reduce pathologically increased diastolic stiffness”.
As evidenced by the clinical relevance of these papers, the insights gained from isolated cardiomyocytes are invaluable – improved understanding of cardiac myocyte function improves our understanding of the function of the heart.