Despite the great amount of research conducted to understand cardiac muscle function and overall cardiovascular health, effective therapies for conditions such as heart failure have yet to be developed. Our latest publication review takes a look at muscle function in the context of diabetes and heart failure, and how these mechanistic findings can help develop targeted therapies in the future.
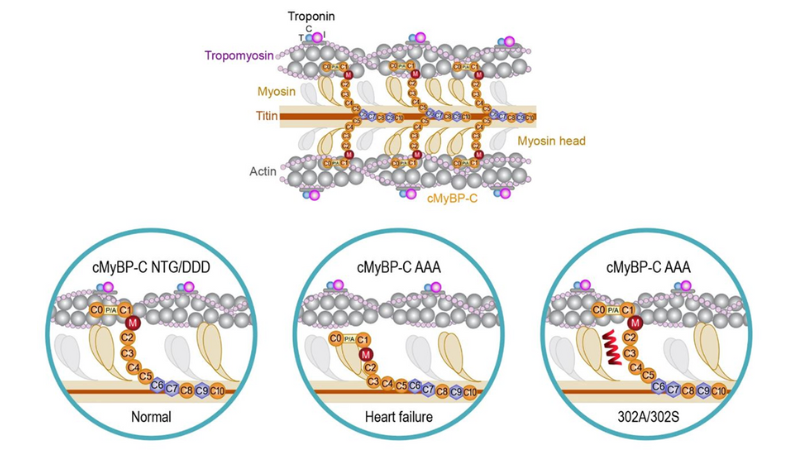
Featured image (© 2022 Hou et al., licensed under CC BY 4.0) illustrates the effect of cardiac myosin binding protein-C (cMyBP-C) phosphorylation state on actomyosin interactions vs. the effect of cMyBP-C peptides 302A and 302S in the context of cross-bridge formation.
Increased Myofilament Calcium Sensitivity is Associated with Decreased Cardiac Troponin I Phosphorylation in the Diabetic Rat Heart
Diabetes impairs systolic and diastolic function, and may predispose individuals to ischemic disease and heart failure. Since changes in calcium availability cannot fully explain this cardiac dysfunction, Greenman et al. explored whether cardiomyocyte myofilament function is altered during the time course of diabetes, as well as the mechanisms underlying changes in calcium sensitivity. For this study, cardiomyocyte force in Zucker Diabetic Fatty (ZDF) rats was measured using our 1600A Permeabilized Myocyte System, while sarcomere length, cell length, and cell height were measured with our 900B Video Sarcomere Length software. Data from this study revealed that ZDF rats have increased calcium sensitivity, though no evidence of left ventricular systolic or diastolic function was found. Additionally, ZDF rats exhibited decreased cardiac troponin I (cTnI) phosphorylation. The authors note that although this study cannot prove causation, these findings indicate that cTnI phosphorylation plays an important role in altering single cell function in the diabetic heart.
Modulation of Myosin by Cardiac Myosin Binding Protein-C Peptides Improves Cardiac Contractility in Ex-Vivo Experimental Heart Failure Models
Cardiac myosin binding protein-C, which regulates sarcomere structure and function in the heart, is highly phosphorylated in normal physiological conditions. However, its phosphorylation levels decrease in diseased states, which has been observed both preclinically and clinically. Dephosphorylated cMyBP-C strongly interacts with myosin, thereby preventing force-generating interaction with actin. Despite the known benefits of targeting this protein, no drug on the market currently improves cardiac function by directly modifying dephosphorylated cMyBP-C. Hou et al. recently tested two peptides in heart failure models in vitro to determine if modulating cMyBP-C dephosphorylation affects cardiac contraction and relaxation. In this study, force measurements were conducted with our 403A Force Transducer and 322C High-Speed Length Controller, while data were acquired and analyzed using our 600A Real-Time Muscle Data Acquisition and Analysis System. The cMyBP-C peptides were shown to disrupt tight myosin-cMyBP-C interactions and improve muscle contractility and ATPase rates in rodent models of heart failure. In contrast, these changes were not observed in control animals, meaning that these effects are specific to the pathological conditions of heart failure. The authors conclude that modulating cMyBP-C dephosphorylation can improve myosin function, sarcomere contractility, and relaxation after a cardiac event, and could complement standard therapies for heart failure.
Reduced Cardiac Muscle Power with Low ATP Simulating Heart Failure
Intracellular ATP levels may be reduced by 50% in patients with heart failure, which has been thought to contribute to impaired myocardial contraction and decreased pump function. Since it is not yet clear whether decreased myocardial ATP is sufficient to impair contractile protein function, Beard et al. recently investigated how low levels of MgATP affect myocardial contraction using demembranated cardiac muscle preparations, and employed our 1400A Permeabilized Fiber Test System. Additionally, our 900B Video Sarcomere Length system was used to observe and measure muscle sarcomeres. The authors found that a 50% decrease in MgATP did not reduce maximum force generation or shortening velocity, but did reduce maximal power generation by 20-25%, and slowed cross-bridge cycling kinetics by 20%. Findings from this study suggest that decreased ATP in heart failure impairs cardiac contraction power and contributes to impaired pump function. The authors conclude that therapies that increase cellular MgATP levels or inhibit adenine nucleotide degradation may be beneficial in treating heart failure.