Models")
Despite recent advances in translational research and general healthcare, Duchenne muscular dystrophy (DMD) remains 100% fatal. This muscle-wasting disease results from a mutation of the dystrophin gene, which is located on the X chromosome – and thus predominantly affects boys. As those born with Duchenne are only likely to reach their mid 20’s, it is clear that a treatment, and ultimately a cure, is of immense importance. Three recent studies have demonstrated that improved treatment is possible, which we discuss in this publication review.
Featured image (©Russell et al. (2023), licensed under CC BY 4.0), shows a compound, EDG-5506, which inhibits the ability of myosin to hydrolyze ATP and develop force by decreasing strong binding between myosin and actin within the sarcomere.
Anti-RANKL Therapy Prevents Glucocorticoid-Induced Bone Loss and Promotes Muscle Function in a Mouse Model of DMD
Bone loss, aside from muscular defects, is one of the key symptoms of this genetic disorder and bisphosphonates are often used as the standard of care to treat this in DMD patients. However, targeting the receptor activator of nuclear factor kappa-B ligand (RANKL) as an alternative, has shown potential to ameliorate muscle dysfunction and also prevent future bone loss. While this could be immensely beneficial for DMD patients, the authors of this paper had to first validate this in suitable Duchenne animal models.
Jayash et al. (2023) assessed the effects of anti-RANKL treatment on skeletal muscle function and bone loss in a variety of mice. Skeletal muscle contractility was measured with Aurora Scientific’s 305B-LR Dual-Mode Lever system in addition to our Dynamic Muscle Control and Analysis system . The authors found that, after an 8-week treatment, anti-RANKL was as effective as other methods of treating DMD, such as deflazacort (DFZ) along and co-treatment of anti-RANKL + DFZ. Notably, anti-RANKL treatment in accompaniment with deflazacort was most effective at improving whole limb grip force in the DMD mice. As the RANK/RANKL/OPG pathway is crucial for bone remodeling, this method of treatment holds tremendous potential as a therapeutic for DMD.
Microdystrophin Gene Addition Significantly Improves Muscle Functionality and Diaphragm Muscle Histopathology in a Fibrotic Mouse Model of DMD
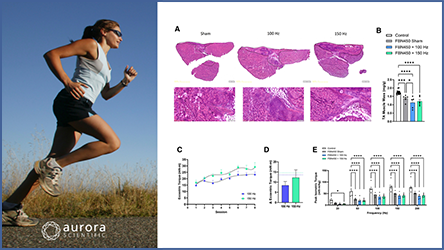
Cernisova et al. (2023) addressed a different facet of DMD, focusing on improving muscle function in the diaphragm using AAV-microdystrophim (AAV8-MD1). As this method of treatment has not been tested in the diaphragm, a muscle crucial to the DMD pathology, understanding its effectiveness in restoring function is of the utmost importance.
After eight weeks of AAV8-MD1 treatment in juvenile Duchenne mice, the authors observed improved muscle mass, decreased fibrotic content, and improved overall histopathology of the diaphragm muscles. Diaphragm function was determined using Aurora Scientific’s 701A simulator and was found to be significantly improved following AAV delivery, in addition to protection from contraction-induced damage, a key hallmark of dystrophic muscle. The administration of AAV8-MD1 also resulted in restored dystrophin expression in the diaphragm. This study provides further support for the use of the MD1 construct as a means to restore dystrophin expression, dramatically improving muscle function and histopathology.
Modulating fast skeletal muscle contraction protects skeletal muscle in animal models of DMD
One other crucial component to the development of DMD is the effect of mechanical stress on muscle dysfunction, as this can lead to exaggerated membrane injury and fiber breakdown, with fast fibers being particularly prone to damage. Muscle contraction, controlled by the motor protein myosin, is a major contributor to these injuries, but the pathophysiology of how muscle contraction and fast muscle fiber damage affects DMD is not well understood.
Russell et al. (2023) sought to further investigate this topic by orally administering an active inhibitor of fast skeletal muscle myosin, known as EDG-5506. Aurora Scientific’s 1400A permeabilized fiber system, as well as our 1200A Isolated Muscle System and 1300A Whole Animal System were used to assess how a reduction in contraction would affect skeletal fibers and whole muscle of DMD mice. Interestingly, only a slight decrease in contraction (<15%) protected dystrophic mice from skeletal muscle injury and fibrosis, with no detrimental effects on strength and coordination. These findings indicate EDG-5506 has a potential to be an alternative treatment strategy for DMD and other related myopathies.
Conclusion
Better treatment options and, ultimately, a cure for Duchenne muscular dystrophy remains a pressing concern, as highlighted by the innovative therapeutic strategies discussed in the reviewed studies. Our specialized instrumentation played a small, yet integral role in generating reliable data, further underscoring the importance of robust scientific tools in this ongoing journey toward improved healthcare outcomes for DMD patients.