The month of March brings with it the promise of Spring and renewal, reflecting a time of growth and cultivation across the natural world. In parallel, researchers across the skeletal muscle field are coming out in full bloom this year for the Advances in Skeletal Muscle Biology conference in sunny, Gainesville, Florida, to discuss the latest and greatest in their research. Spanning the topics of muscle atrophy, growth, and regeneration, as well as exercise, aging, bioenergetics and more, there’s no shortage of topics to delve into. In fact, recent discoveries have sprouted a particular interest in protein perturbations to skeletal muscle health. The following publication review covers the impacts of protein dysfunctions, knockdowns, and modifications.
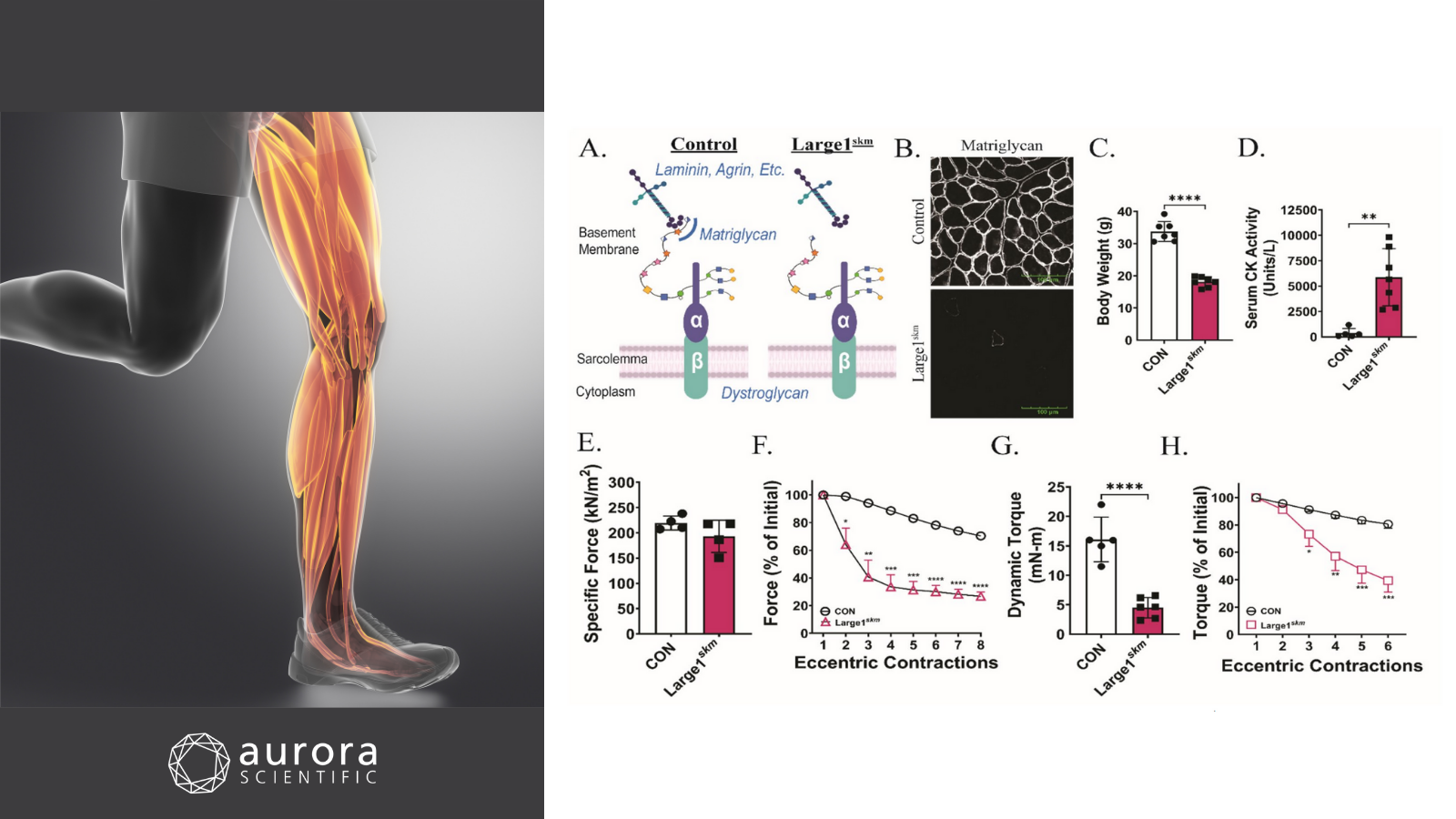
Featured image with a photo of leg muscles (©Janulla from Getty Images via Canva.com) and figures (adapted from ©Hord et al. (2025), licensed under CC BY 4.0) depicting how skeletal muscle health requires matriglycan. A) Schematic of DG core M glycans in control and Large1skm mice, B) immunofluorescence of skeletal muscle matriglycan, C) body weights of 20-week-old control and Large1skm mice, D) serum creatine kinase (CK) activity two hours after treadmill exercise, E) specific force of maximum isometric tetanic contractions in ex-vivo EDL muscle, F) eccentric contractions in ex-vivo EDL muscle, G) in-vivo dynamic torque production in plantar flexors, and H) isometric torque in plantar flexors after repeated eccentric contractions.
Sarcolemma resilience and skeletal muscle health require O-mannosylation of dystroglycan
Skeletal muscle function relies on the critical linkage between the extracellular matrix (ECM) and the sarcolemma, a connection mediated by dystroglycan (DG). For DG to bind the ECM, it undergoes O-mannosylation, initiated by the protein O-mannosyltransferase 1 (POMT1) and protein O-mannosyltransferase 2 (POMT2) enzyme complex. This in turn produces three glycan subtypes: core M1, M2, and M3, in which the M3 glycan is capped with matriglycan. While mutations in POMT1 are known to underly severe muscular dystrophies such as limb-girdle muscular dystrophy (LGMB) and Walker-Warburg Syndrome (WWS), how both O-mannosylated DG and matriglycan impact muscle health remain markedly unclear. To uncover this, Hord et al. (2025) generated and characterized skeletal muscle-specific Pomt1 knockout (Pomt1skm) mice as well as mice that lacked matriglycan (conditional deletion of like-acetylglucosaminyltransferase 1; Large1skm).
For phenotypic assessment of the mice, body weight, grip testing, hanging test, open-field, in-vivo torque and ex-vivo muscle force experiments were conducted. Aurora Scientific’s 1300A 3-in-1 Whole Animal System was used to measure plantar flexion of the triceps surae. To achieve this, electrodes were subcutaneously placed in proximity to the tibial nerve, followed by isometric tetanic contractions and dynamic eccentric contractions. To further measure ex-vivo skeletal muscle contractile properties, the extensor digitorum longus (EDL) was carefully dissected and assessed with the 1200A: Isolated Muscle Test System by conducting isometric tetani and eccentric contraction protocols.
On the ex-vivo level, maximal isometric tetanic contractile forces were not significantly lower in Pomt1skm or Large1skm muscles compared to controls. Conversely, the in-vivo tests revealed significantly lower body weights and grip strength, shorter travel distances, and significantly lower torque outputs in both knockout mice. Moreover, Pomt1skm and Large1skm muscles were unable to maintain force production after repeated lengthening contractions, and the plantar flexors were highly susceptible to eccentric contraction-induced damaged. To rescue these phenotypes in Pomt1skm mice, gene transfer of Pomt1 was performed via administration of adeno-associated virus (AAV)-Pomt1. Although body weights were no different, treated mice exhibited significantly higher grip strength and torque, with control-like responses to eccentric contractions at 16-weeks. These results remarkably illustrate the importance of both O-mannosylated DG and matriglycan in neuromuscular function and demonstrate how genetic rescue of Pomt1 can promisingly limit skeletal muscle pathology.
Defective cystic fibrosis transmembrane conductance regulator accelerates skeletal muscle aging by impairing autophagy/myogenesis
Cystic fibrosis (CF) is a progressive, genetic disease, caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, located on chromosome 7. These mutations lead to dysfunction of the CFTR protein, a critical chloride channel located on the surface of cells. While CF is known to cause frequent respiratory infections, airway issues, and shortness of breath, peripheral muscle atrophy and weakness are also notably prevalent in patients. Interestingly, despite its association with muscle health, the impact of CFTR on skeletal muscle regeneration and aging is unknown. To address this, Chen et al. (2025) examined CFTR expression across human samples and mouse models, with a specific focus on skeletal muscle myogenesis and aging.
The expression of the CFTR gene was assessed via quantitative PCR (qPCR) analysis in human biopsy samples, aged mice, and senescence-resistant mice. This was followed by ex-vivo muscle function tests using Aurora Scientific’s 800A: in-vitro Muscle Apparatus and 610A: Dynamic Muscle Control software. Isometric twitch contractions, tetanic contractions, and fatiguability were assessed in the gastrocnemius of wildtype and CFTR-deficient mice.
Intriguingly, CFTR was found to be age-dependently downregulated in both the human biopsy samples and aged mice, suggesting that its expression in skeletal muscles decreases with age. Moreover, CFTR-deficient mice presented with age-dependent skeletal muscle defects, exhibiting decreased specific twitch and tetanic forces with age. Specifically, a significantly lower twitch force was observed in 18-month-old CFTR-deficient mice compared to wildtype controls. In myoblast cultures from CFTR-deficient mice, defective myogenic differentiation was observed, but was subsequently rescued by overexpressing LC3-β, an autophagy mediator. By the same token, overexpressing CFTR in aged mice increased mitochondrial LC3-β levels and improved skeletal muscle mass and function. Taken together, these findings demonstrate how CFTR expression could serve as a potential treatment strategy for age-related skeletal muscle defects.
Inducible and reversible SOD2 knockdown in mouse skeletal muscle drives impaired pyruvate oxidation and reduced metabolic flexibility
Mitochondria are comically renown as the “powerhouses of the cell” within the collective conscious of science students alike. Despite this widely remembered definition, the specific role of skeletal muscle mitochondria in maintaining muscle function and metabolic regulation is less known. In fact, mitochondrial dysfunction and oxidative stress are cited hallmarks of skeletal muscle aging and disease, raising questions on whether oxidative stress is a cause or a consequence of this phenomenon. To delineate this, Ostrom et al. (2025) developed an inducible and reversible skeletal muscle knockdown of superoxide dismutase 2 (SOD2), an antioxidant enzyme responsible for the conversion of superoxide to hydrogen peroxide. This model, coined iSOD2 KD, works by leveraging a tetracycline-controlled skeletal muscle specific short hairpin RNA (shRNA) targeted to Sod2 mRNA. In essence, doxycycline (DOX) treatment in the iSOD2 KD model leads to the degradation of Sod2 mRNA in skeletal muscles, allowing for precise control of mitochondrial oxidative stress in the muscles of adult mice.
To characterize the KD model, mice were administered DOX treatment for 3- or 12-weeks and followed for up to 24 weeks. Across these timepoints, genotyping, western blotting, aconitase activity measurements, high resolution respirometry and muscle function tests were collectively performed. In particular, Aurora Scientific’s 1300A 3-in-1 Whole Animal System was employed to conduct force frequency and fatigue tests in both control and KD mice following 8 weeks of continuous DOX treatment.
Upon analysis, the 12-week DOX treatment revealed successful knockdown of SOD2 in the gastrocnemius, soleus, tibialis anterior, quadriceps and heart tissue, without changes in the liver, kidney or brain. At the 8-week timepoint, there were no significant differences in the force frequencies of control and KD mice, but the KD muscle exhibited a small, but significantly reduced, fatigue resistance compared to controls. Moreover, complex I-driven respiration and maximal oxidative phosphorylation capacity were reduced, particularly at 12- and 20-weeks post-recovery from DOX. Mitochondrial aconitase activity awas also significantly decreased in muscle of KD mice, with the largest reduction occurring at week 12 post-DOX treatment. In effect, this new model underscores the impact of elevated matrix superoxide, serving as a useful investigative tool for skeletal muscle metabolism and redox biology studies.
Conclusions
Springing up as our second exhibiting conference of 2025, the Advances in Skeletal Muscle Biology conference is always one of our favourites in the field. While Aurora Scientific continues to network and learn from the experts on the ground, these publications by Hord et al. (2025), Chen et al. (2024), and Ostrom et al. (2024) advance our understanding of protein perturbations in skeletal muscle biology.


