Muscular dystrophy (MD), a group of incurable diseases, is caused by genetic mutations that lead to progressive muscle weakness and loss of muscle mass. Preclinical models have greatly advanced our understanding of the molecular mechanisms that underlie MD and have paved the way for novel therapeutic approaches that have potential for clinical translation. Our muscle physiology products have been used in a variety of preclinical applications, and have recently been employed in studies of Duchenne MD (DMD), Limb-girdle MD (LGMD), and oculopharyngeal MD (OMPD), which are summarized below in this publication review.
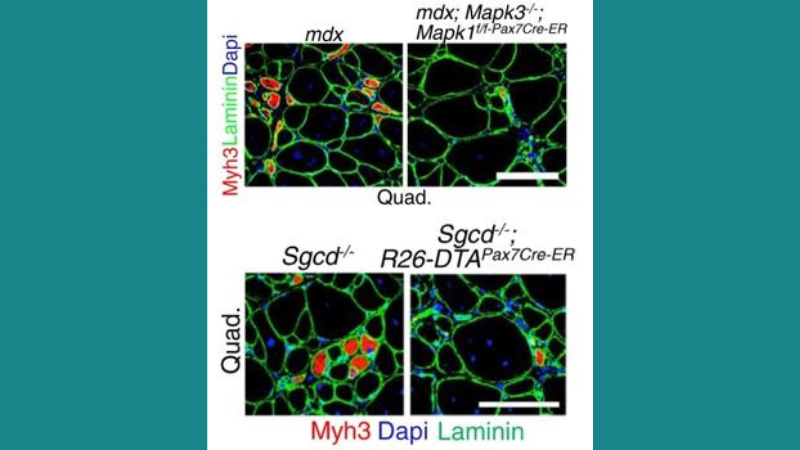
Featured image (© 2022, Boyer, J. G. et al.) highlights representative quadricep muscle sections immunostained for Myh3 (red) and laminin (green) in four-month-old mice of the indicated genotype, with nuclei stained by Dapi (blue). Scale bars represent 100 µm.
Depletion of Skeletal Muscle Satellite Cells Attenuates Pathology in Muscular Dystrophy
Resident progenitor “satellite” cells can repair damaged muscle fibers and replace them following death, and are currently being investigated as an MD treatment. Recently, Boyer et al. assessed how satellite cells contribute to MD pathogenesis in mdx and Sgc-/- mice, which are models of DMD and LGMD type 2F, respectively, and employed our 305C muscle lever system for muscle performance measurements. Results from these experiments demonstrated that satellite cell depletion improved MD histopathology and muscle performance in these mice, and reduced muscle susceptibility to contraction injury. The authors also mechanistically demonstrated that chronic satellite cell activity induces the re-expression of the myogenic transcription factor MyoD and the fetal gene program in muscle fiber, which destabilizes the sarcolemma. Although depleting satellite cells in MD patients is not currently recommended, the authors concluded that limiting satellite cell activity could be effective in mitigating MD pathology.
Pharmacological Activation of SERCA Ameliorates Dystrophic Phenotypes in Dystrophin-Deficient mdx Mice
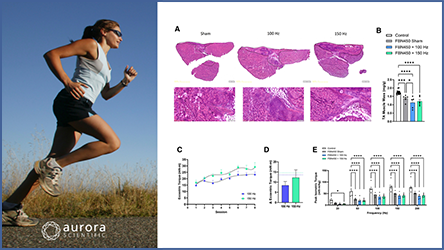
Calcium regulation by sarco/endoplasmic reticulum Ca2+ -ATPase (SERCA) has been shown to be chronically impaired in the sarcoplasmic reticulum of DMD mouse dystrophic muscle. Previous reports have demonstrated that CDN1163, an allosteric SERCA activator, may be applicable for DMD treatment. In this study, Nogami et al. further investigated the therapeutic potential of CDN1163 in mdx mice, and utilized our 1200A in-vitro muscle test system to perform muscle twitch and tetanic force measurements. The authors demonstrated that pharmacological SERCA activation could decrease cytosolic Ca2+ levels in mdx mice and attenuate dystrophic phenotypes. One week of CDN1163 treatment restored mitochondrial function in these mice and prevented muscular damage caused by exercise, while seven weeks of treatment reduced muscular degeneration and improved muscular strength. The authors concluded that activating SERCA by CDN1163 administration could be a promising therapeutic strategy for DMD, but its effects on cardiac muscle remain to be assessed.
BB-301: A Silence and Replace AAV-Based Vector for the Treatment of Oculopharyngeal Muscular Dystrophy
OPMD is a rare neuromuscular disorder characterized by ptosis and dysphagia-related complications. Although Strings-Ufombah et al. have previously demonstrated the efficacy a two-vector adeno-associated virus-based gene therapy approach in a murine OPMD model, translation to humans is limited by high manufacturing costs and risks, as well as regulatory challenges. The authors have therefore combined the essential elements of the two initial vectors into BB-301, a single “silence and replace” recombinant vector, and measured in-situ muscle force in A17 mice with our 1300A whole animal system. Experimental findings indicated that intramuscular injection of BB-301 into tibialis anterior muscle improved disease pathology and muscle function. Furthermore, suboptimal BB-301 doses also fully restored muscle strength and weight, but at a slower rate than higher doses. Currently, additional preclinical studies are underway to investigate the local administration of BB-301 into muscles of the pharyngeal complex. The authors also noted that this therapy has received orphan drug designation by the FDA and is being prepared for administration in human OMPD patients.