Diabetes mellitus (DM), commonly referred to as diabetes, is a condition where the body loses its ability to regulate blood glucose levels. It is primarily classified into Type 1 diabetes (T1DM), which involves impaired insulin production, and Type 2 diabetes (T2DM), which is characterized by progressive insulin resistance. Over the years, diabetes has been the subject of growing global concern due to its effects on muscle, especially in the heart. In the following publication review, we delve into the latest discoveries in diabetes research, shedding light on its wide-reaching impact on muscle health and overall well-being.
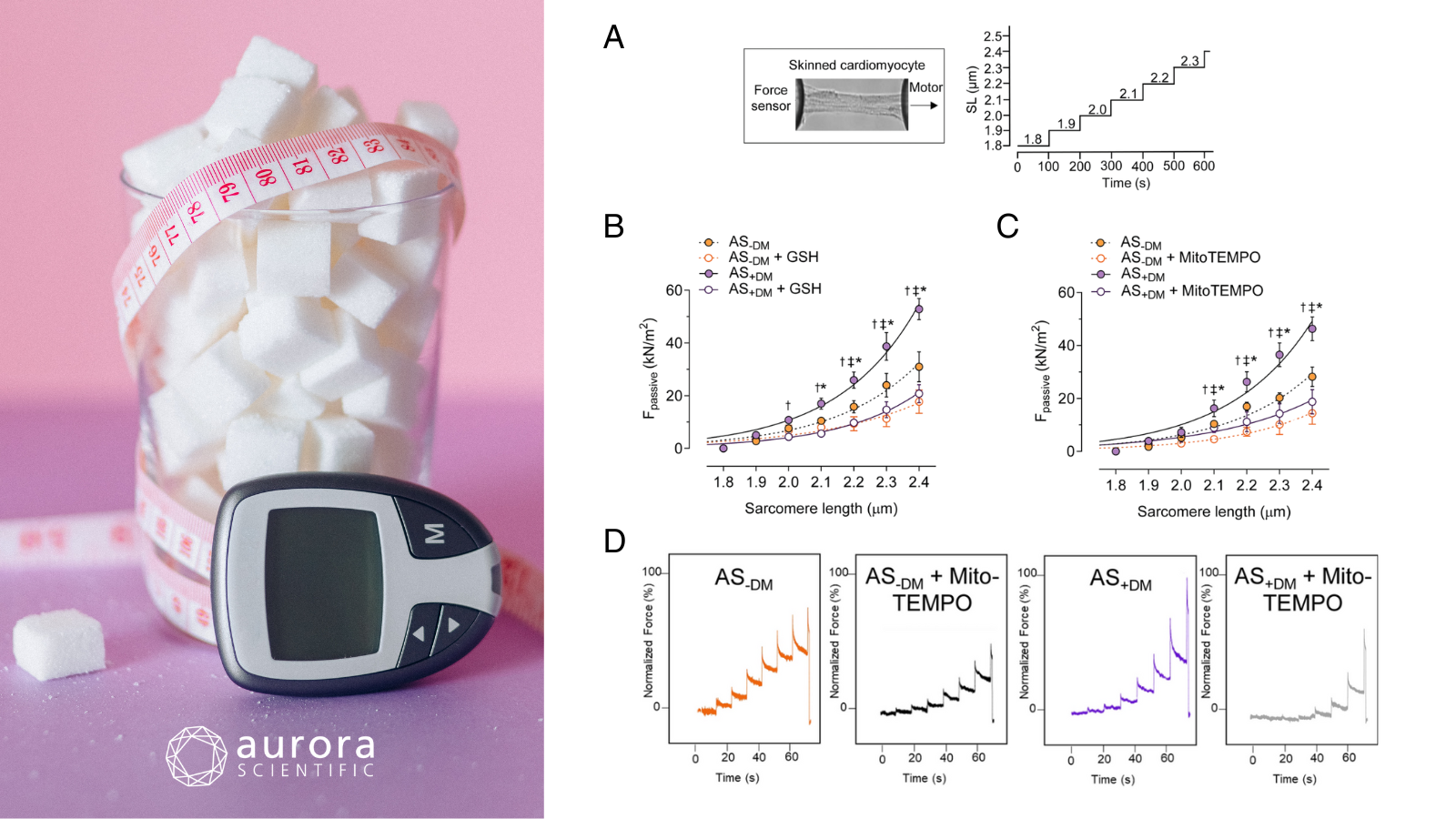
Featured image with sugar cubes in a glass (©Nataliya Vaitkevich via Canva.com) and figures (adapted from ©Herwig et al. (2025), licensed under CC BY 4.0) depicting A) a skinned cardiomyocyte stretch protocol, where SL refers to sarcomere length, B) passive force at sarcomere length 1.8-2.4 µm in the presence or absence of reduced glutathione, C) passive force at sarcomere length 1.8-2.4 µm in the presence or absence of MitoTEMPO, and D) original force measurement recordings.
Diabetes mellitus aggravates myocardial inflammation and oxidative stress in aortic stenosis: a mechanistic link to HFpEF features
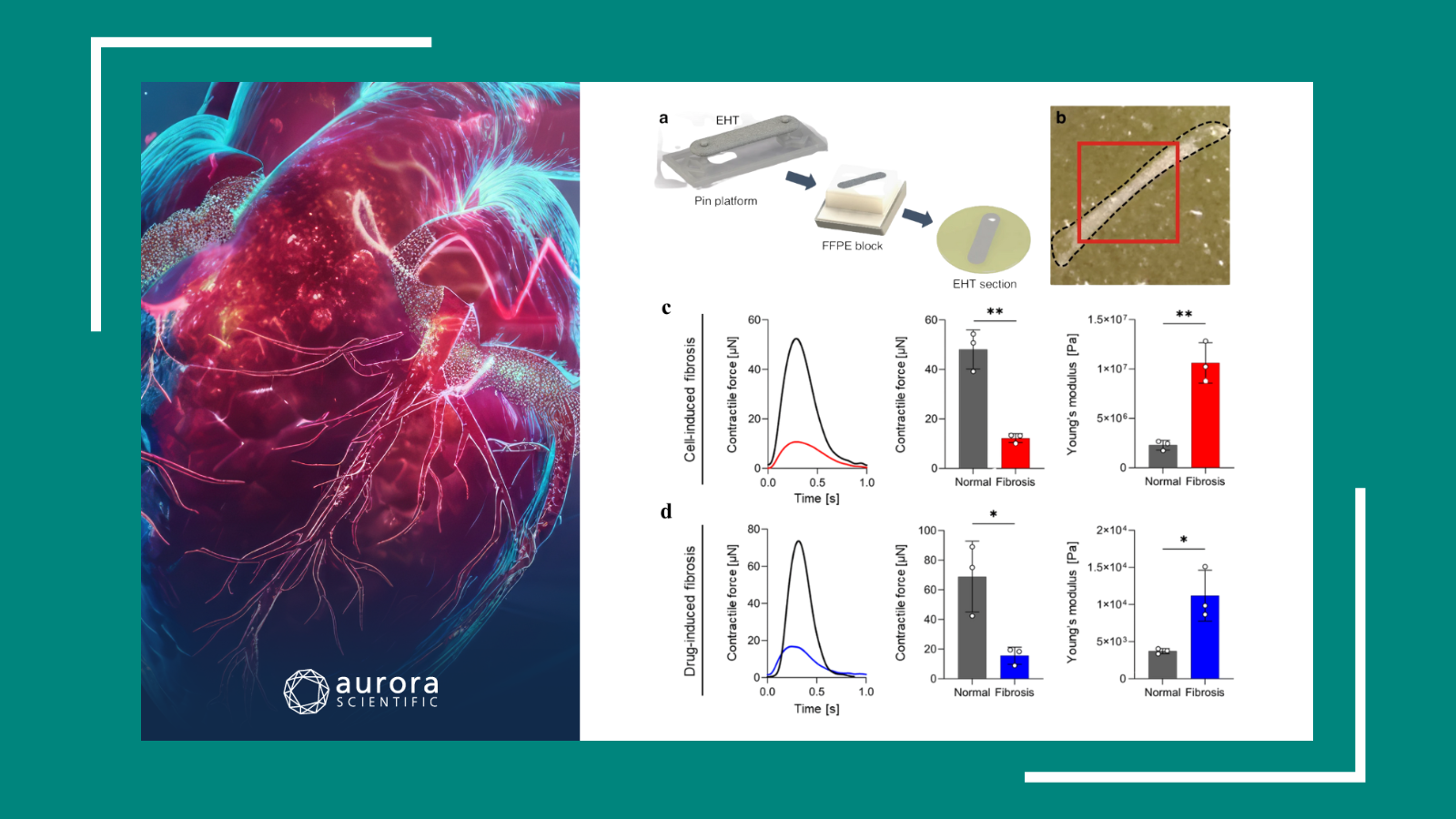
Diabetes mellitus (DM) is known to exacerbate outcomes in patients with aortic stenosis (AS), a condition characterized by the narrowing of the aortic valve opening. Beyond accelerating AS progression, DM also significantly raises the risk of cardiovascular mortality and heart failure hospitalizations. However, the underlying mechanisms through which DM contributes to these complications remain poorly understood. To address this gap, Herwig et al. (2025) explored the impact of DM on the left ventricle in AS patients.
Myocardial biopsies were collected from 28 diabetic AS patients and compared with those from 17 non-diabetic AS patients. Aurora Scientific’s 1600A: Permeabilized Myocyte System and 403A: Force Transducer were employed to measure the passive force in permeabilized cardiomyocytes. In addition, western blotting, ELISA, molecular assays, echocardiography, and histology experiments were performed to assess inflammatory responses and tissue changes.
Upon measurement, cardiomyocytes from the diabetic cohort exhibited increased passive stiffness compared to non-diabetic AS patients. Biochemical and molecular analyses further revealed elevated levels of pro-inflammatory mediators, oxidative stress markers, and interleukins, suggesting heightened inflammation and oxidative stress in these patients. Interestingly, treatment with an IL-6 inhibitor and the antioxidant glutathione (GSH) ameliorated these alterations, normalizing the passive stiffness to levels seen in the non-diabetic group. Similarly, treatment with protein kinase G (PKG) and the sodium-glucose cotransporter 2 (SGLT2) inhibitor empagliflozin also reduced passive stiffness in diabetic AS patient cardiomyocytes, though not to the degree observed in non-diabetic AS patients. As such, this study highlights the exacerbating effects of diabetes on AS pathology and offers promising therapeutic strategies for further investigation.
Dysregulated inflammation, oxidative stress, and protein quality control in diabetic HFpEF: unraveling mechanisms and therapeutic targets
Type 2 diabetes mellitus (T2DM) is a major risk factor for cardiovascular disease and is strong linked to the development of heart failure with preserved ejection fraction (HFpEF). As the prevalence of T2DM continues to rise and HFpEF emerges as the most common form of heart failure, the interplay between these conditions represents an escalating global health concern. While systemic metabolic dysfunction in diabetic HFpEF has been increasingly recognized, the specific physiological mechanisms driving disease progression remain poorly understood. To address this, Delalat et al. (2025) explored how T2DM-related disruptions affect metabolic, inflammatory, and oxidative stress pathways, as well as cardiomyocyte stiffness in HFpEF.
Left ventricular biopsies were obtained from HFpEF patients with and without T2DM and subjected to a range of molecular, biochemical, and biomechanical analyses. Passive force measurements were performed on single, skinned cardiomyocytes using Aurora Scientific’s 1600A: Permeabilized Myocyte System, equipped with a 403A Force Transducer and 315D High-Speed Length Controller, to assess sarcomere-length-dependent stiffness. Additional analyses included immunofluorescence imaging, Western blotting, ELISAs, and qRT-PCR to evaluate inflammation, oxidative stress, and signaling pathways involved in myocardial dysfunction.
During the force measurements, cardiomyocytes from diabetic HFpEF patients were significantly stiffer than those from non-diabetic patients, particularly at key resting sarcomere lengths. This heightened stiffness was largely driven by elevated inflammation, including IL-6, and was significantly reduced with IL-6 inhibition, revealing a critical connection between inflammatory signaling and impaired cardiac mechanics. Oxidative stress and decreased levels of protective heat shock proteins (HSP27 and HSP70) further exacerbated stiffness; however, both antioxidant therapy and HSP supplementation effectively improved cardiomyocyte mechanical properties. Together, these findings highlight the central roles of inflammation, oxidative stress, and disrupted protein quality control (PQC) in driving cardiomyocyte stiffness in diabetic HFpEF, pointing to promising therapeutic targets for this high-risk group.
Targeting Myostatin as an Adjunct Treatment for the Preservation of Cardiometabolic and Skeletal Muscle Function in Type 1 Diabetes
Type 1 diabetes mellitus (T1DM) is a chronic autoimmune disease marked by insulin deficiency, hyperglycemia, and progressive skeletal muscle and vascular dysfunction. While exercise can help mitigate these effects by promoting muscle growth and improving glucose uptake, many T1DM patients are unable to maintain sufficient physical activity to gain these benefits. Myostatin, a negative regulator of muscle growth that is elevated in T1D, presents a promising target for addressing both muscle wasting and metabolic dysfunction. Nunan et al. (2025) aimed to evaluate whether myostatin deletion could preserve muscle mass and function, improve glucose regulation, and protect vascular health in a mouse model of T1D.
Adult male mice with and without genetic deletion of myostatin were used to assess the impact of myostatin on T1D-induced cardiometabolic dysfunction. T1DM was chemically induced using low-dose streptozotocin, and glucose homeostasis, adiposity, metabolism, vascular, and muscle function were evaluated. Gastrocnemius muscle performance was assessed in-vivo using Aurora Scientific’s 1300A Whole Animal System, which allowed for precise measurement twitch, force-frequency, and fatigue force testing.
Deletion of myostatin was found to preserve glucose homeostasis by preventing hyperglycemia, improving glucose tolerance, and reducing HbA1c levels, despite insulin being undetectable in all STZ-treated mice. Myostatin knockout also protected against T1DM-induced impairments in skeletal muscle function, preserving isometric force production across a range of frequencies. Additionally, myostatin deletion prevented endothelial dysfunction in skeletal muscle arterioles and attenuated classic signs of metabolic dysfunction such as hyperphagia, polydipsia, and polyuria. These results demonstrate that targeting myostatin may serve as a promising adjunct therapy to preserve cardiometabolic and skeletal muscle health in T1DM, even in the absence of insulin.
Conclusions
These diabetes discoveries and determinations by Herwig et al. (2025), Delalat et al. (2025), and Nunan et al. (2025) drive home the critical impact of DM on muscle health. Covering insights from inflammation to oxidative stress and muscle dysfunction, they offer mechanistic insights and therapeutic avenues that hold promise for improving cardiovascular, metabolic, and skeletal muscle health across diabetes.