The cardiovascular sciences intersect with muscle physiology, molecular genetics, pharmacology, stem cell applications and more, marrying a multivariate stream of fields. For this reason, the Basic Cardiovascular Sciences meeting is held annually across the United States, offering a venue to explore, share, and collaborate on the latest cardiovascular insights. Without missing a beat, the following publication review encapsulates the breadth of the conference, covering a novel limb function test for preclinical peripheral artery disease models, a promising drug treatment for genetic cardiomyopathies, and contributions of myofilament networks to passive stiffness.
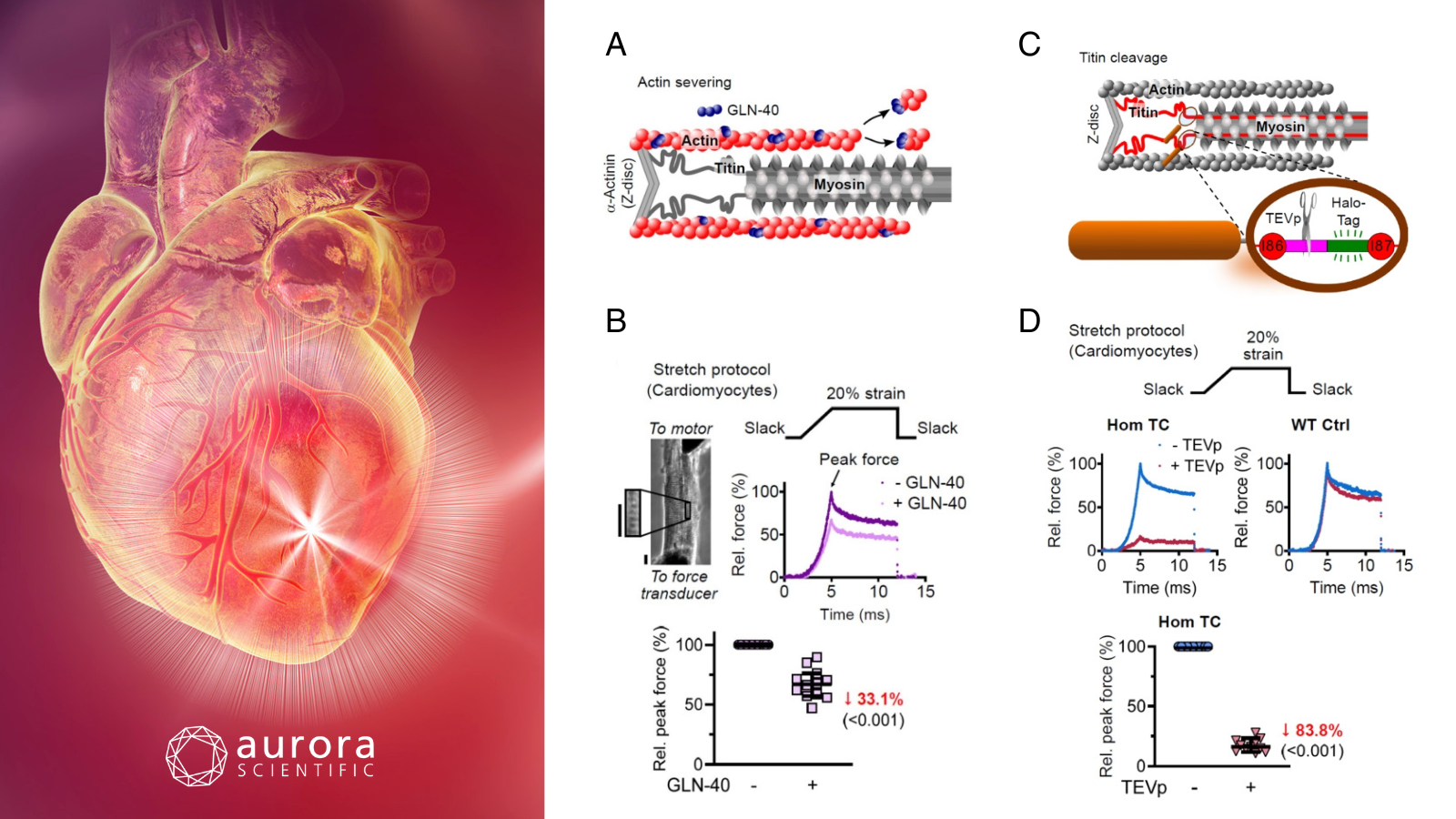
Featured image with a conceptual computer illustrated heart attack (©Science Photo Library via Canva.com) and figures (adapted from ©Wagner et al. (2025), licensed under CC BY 4.0) depicting the contribution of actin filaments and titin springs to transverse and longitudinal passive stiffness. A) Schematic of actin severing by GLN-40, B) passive force in permeabilized cardiomyocytes before and after GLN-40 treatment; including the stretch protocol, phase image with ROI, representative force traces, and normalized peak force, C) schematic of TEVp cleavage site in titin’s I-band (Hom TC model), and D) passive force in cardiomyocytes before and after TEVp treatment, showing stretch protocol, force traces (Hom TC and WT), and normalized peak force.
A 6-Minute Limb Function Assessment for Therapeutic Testing in Experimental Peripheral Artery Disease Models
Peripheral artery disease (PAD) is a vascular disorder leading to the abnormal narrowing of arteries in the arms and legs. Although PAD affects over 230 million people globally and increases the risk for cardiovascular events, therapeutic progress has remained stagnant for decades. A key issue underlying the gap between preclinical therapies and their translation to the clinic is the contrasting methodology for assessing therapeutic efficacy. While preclinical models rely on limb perfusion recovery, a 6-minute walk test is employed clinically. To bridge this gap, Palzkill et al. (2025) developed a translational 6-minute limb function test for the assessment of muscle perfusion, power, and work output in preclinical models.
Aurora Scientific’s 1300A: Whole Animal System was used to measure isometric and isotonic muscle function in mice with hindlimb ischemia. The sciatic nerve was electrically stimulated to induce contractions in the plantar flexors, with force measured via the 300C: Dual-Mode Muscle Lever. For the 6-minute limb function test, isotonic contractions were triggered under controlled load conditions to assess performance metrics such as force, velocity, power, and work.
Upon testing, the protocol effectively distinguished differences in limb function between genetically distinct mouse strains, detected improvements following exercise training, and revealed enhanced muscular work capacity in mice overexpressing PGC1α, a regulator of mitochondrial function. This novel test bridges the gap between preclinical and clinical PAD assessments by mimicking functional outcomes like walking ability, providing a powerful tool to evaluate therapeutic interventions targeting both muscle and vascular deficits
An FDA-approved drug structurally and phenotypically corrects the K210del mutation in genetic cardiomyopathy models
Dilated cardiomyopathy (DCM) is a heart disease whereby the heart chambers enlarge and lose their ability to contract. In particular, DCMs arising from genetic mutations remain poorly understood at the structural level, limiting the development of targeted therapies. Among these, the K210del mutation is a well-established cause of familial DCM; however, its mechanistic impact on calcium handling and myocardial function has not been structurally defined. Therefore, Wang et al. (2025) aimed to resolve the crystal structure of the K210del troponin complex and explore structure-based therapeutic correction using a repurposed FDA-approved drug.
A multidisciplinary approach combining structural biology, in silico modeling, cell biology, and in vivo physiology were employed. Crystal structures of both wild-type and K210del mutant troponin complexes were resolved, followed by structure-based drug screening and molecular dynamic simulations to identify risedronate as a candidate structural corrector. Functional correction was validated using patient-derived human induced pluripotent stem cell (hiPSC) cardiomyocytes and skinned cardiac papillary muscle fibers from K210del mice. In the latter, Aurora Scientific’s 322C High-Speed Length Controller and 403A Force Transducer were instrumental in quantifying calcium-dependent force generation.
Structural analyses revealed that the K210del mutation allosterically distorts the activation Ca²⁺-binding domain in troponin C by altering local electrostatic surfaces and hydrogen bonding networks. Using structure-based drug screening, FDA-approved risedronate was identified as a corrective agent, restoring the troponin complex’s Ca²⁺-binding configuration and normalizing contractility in patient-derived cardiomyocytes and K210del mouse models. In-vivo, risedronate significantly improved calcium sensitivity and left ventricular function in K210del mice, with effects persisting across different stages of disease and dosing regimens. These findings uncover the structural mechanism underlying calcium desensitization in K210del mutation and identify risedronate as a promising repurposed therapeutic agent capable of reversing the associated contractile dysfunction.
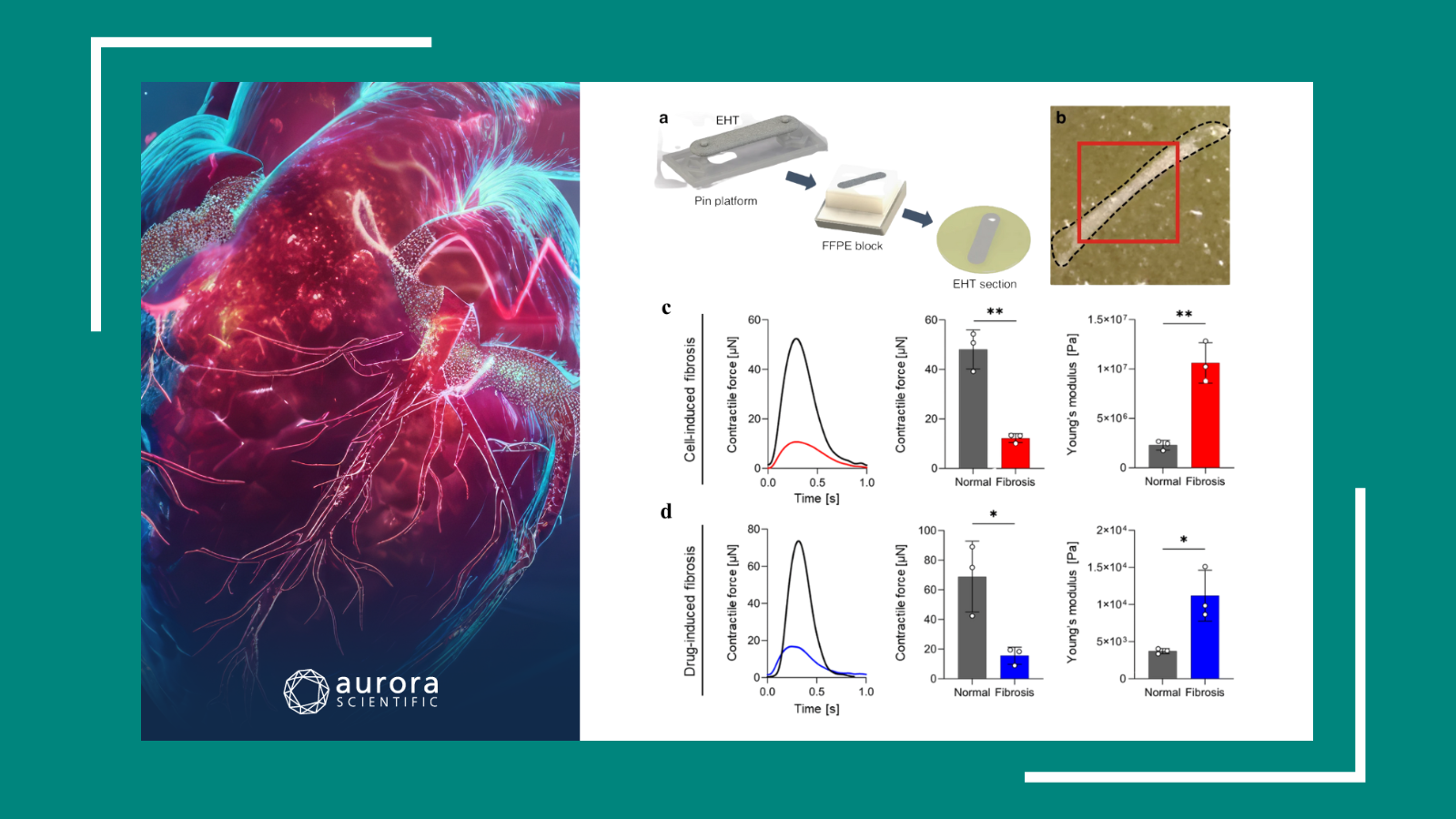
Direction‑dependent contributions of cardiac myofilament networks to myocardial passive stiffness reveal a major disparity for titin
Altered passive stiffness is a hallmark of heart failure, influenced by both extracellular matrix (ECM) components and intracellular myofilament proteins, particularly titin. While longitudinal myocardial stiffness has been well-studied, the mechanical contributions of myofilaments to transverse stiffness remain poorly understood. Wagner et al. (2025) addressed this gap by quantifying the directional roles of titin, actin, and myosin in myocardial passive stiffness using targeted filament disruption and advanced mechanical assays.
Transverse stiffness was assessed in cardiac tissue sections using atomic force microscopy (AFM) nanoindentation, while longitudinal stiffness was measured in single cardiomyocytes and fiber bundles using Aurora Scientific’s 403A Force Transducer. Tissue preparation included targeted disruption of myofilaments, including titin via TEVp protease, actin via GLN-40 gelsolin fragment, and myosin via high-salt extraction. The orchestrated integration of AFM, custom stretching assays, and confocal microscopy allowed for high-resolution, direction-specific mechanical analysis of mouse myocardial tissue.
Titin emerged as the dominant determinant of myocardial elasticity, accounting for over 50% of elastic force at moderate strain and 46–78% of both elastic and viscous forces, depending on the presence of the ECM. Actin and microtubules also contributed significantly, particularly to viscous forces at low strain, while the sarcolemma and ECM played minor but strain-dependent roles. Notably, titin and actin form a non-reciprocal tensegrity system, whereby actin supports titin’s mechanical role, but not vice versa. This comprehensive analysis establishes titin as the principal mechanical scaffold of cardiac passive stiffness, while revealing a dynamic interplay between cytoskeletal and extracellular components that adaptively distribute load in response to strain.
Conclusions
Reflecting the heart of the multifaceted BCVS conference, these studies by Palzkill et al. (2025), Wang et al. (2025), and Wagner et al. (2025) showcase the extensive applications of cardiovascular research. Spanning the development of a novel preclinical test, identification of a promising drug therapy, and delineation of passive stiffness, they further drive our understanding of the multidisciplinary cardiac field.